






一、前言
环氧树脂胶粘剂是以环氧树脂为主体配制而成的。树脂大分子末端有环氧基,链间有羟基和醚键.并在固化过程中还会继续产生羟基和醚键,结构中含有苯环和杂环。这些结构决定了环氧树脂胶粘剂具有如下一些特点:
(1)粘接力大,粘接强度高;
(2)收缩率小,尺寸稳定。环氧树脂胶在固化时几乎不放出低分子产物.线膨胀系数受温度影响小,因此,粘接件的尺寸稳定性好。
(3)电性能优良,耐介质好。环氧树脂胶的固化产物具有优异的电绝缘性能,体积电阻率为1013—1016Ω.cm.介电强度为30—50KV.Mm-1环氧树脂分子中含有醚键,且分子链间排列紧密,交联密度又大,故有良好的耐溶剂、耐油、耐酸、耐碱、耐水等性能,特别是耐碱性强。
(4)易于改性,用途广泛。环氧树脂与很多的橡胶(弹性体)及热塑性树脂相溶性好,甚至发生化学反应;与填料分散性好,可在很大范围内改变环氧树脂胶的性能。
(5)工艺性好,使用方便。
(6)毒性较低,危害性小。
(7)韧性不佳,脆性较大。树脂中含有很多的苯环和杂环,分子链柔性小,加之固化后的交联结构不宜变形.未增韧的环氧树脂胶韧性不好.脆性较大,剥离强度很低,不耐冲击振动。
由此可见,环氧树脂具有良好的综合力学性能,特别是高度的粘合力、很小的收缩率、很好的稳定性、优异的电绝缘性能,作为粘合剂、复合材料基质、粉末涂料等制品提供了物质基础。
三、环氧树脂的增韧研究
过去,人们对环氧树脂的改性一直局限于橡胶方面,如端羧基丁腈橡胶、端羟基丁腈橡胶、聚硫橡胶等。近年来,对环氧树脂的改性不断深入,改性方法日新月异,如互穿网络法、化学共聚法等,尤其是液晶增韧法和纳米粒子增韧法更是近年来研究的热点。归纳起来增韧途径主要有四种:1)在环氧基体中加入橡胶弹性体、热塑性树脂或液晶聚合物等分散相来增韧。2)用热固性树脂连续贯穿于环氧树脂网络中形成互穿、半互穿网络结构来增韧。3)用含有“柔性链段”的固化剂固化环氧,在交联网络中引入柔性链段。提高网链分子的柔顺性。达到增韧的目的。4)刚性粒子与纳米粒子增韧。
1、橡胶弹性体增韧
橡胶弹性体通过其活性端基(如羧基、羟基、氨基)与环氧树脂中的活性基团(如环氧基、羟基等)反应形成嵌段;正确控制反应性橡胶在环氧树脂体系中的相分离过程是增韧成功的关键。
1.1液体羧基丁腈橡胶增韧
在活性端基液体橡胶增韧剂中,研究较早较多的是CTBN。在CTBN/EP体系中,当CTBN的质量分数为l0%时,固化物的断裂韧性(Kc)为2.34MN-m,比未改性的环氧树脂增加了270%。影响CTBN增韧效果的主要因素有CTBN中丙烯腈含量.CTBN的分子质量、添加量,固化剂,固化温度,环氧基体平均网链长度及官能团数目等。CTBN对常温固化体系增韧效果较差。采用交联橡胶粒子改性环氧树脂,橡胶用量为5%一l0%。便可达到剪切强度和剥离强度的平衡,即使在常温下快速固化,也具有很高的剥离强度?冲击强度和很好的粘接耐久性.可用作粘接油面钢板的结构胶粘剂。
液体羧基丁腈橡胶无论是无规羧基液体丁腈橡胶(CBN),还是端羧基液体丁腈橡胶(CTBN),在高温及催化剂存在下,都能与环氧基反应,而把橡胶柔性链段引入环氧树脂交联结构中,起到增韧作用。其中以端羧基液体丁腈橡胶对环氧树脂的增韧效果最好。它能与环氧树脂混溶,在固化过程中形成环氧树脂为连续相,丁腈橡胶为粒子状分散相的两相体系,即所谓的“海岛”式两相结构.获得了显著的增韧效果。CTBN带有可与环氧基发生化学反应的官能团,因此在两相界面上存在化学键结合的强相互作用,使环氧树脂的韧性成倍提高。
以液体羧基丁腈橡胶改性环氧树脂。通常先与环氧树脂进行预反应,以三苯基膦为催化剂.在100-150℃下反应1~2h。形成两端有双酚A环氧树脂链节单元的嵌段或共聚物,再与固化剂形成胶粘剂。活性端基液体橡胶增韧环氧树脂虽然是一条可行的改性途径,但价格比较昂贵,目前主要在军事工业上应用,在民用领域还未规模化使用。端羟基丁腈橡胶(HTBN)液体橡胶价格相对便宜.与环氧树脂相容性好,增韧效果与CTBN相当.通过进一步改进性能,可望在环氧树脂改性中发挥重要作用。端羧基丁腈橡胶(CTBN)能使环氧树脂显著提高断裂韧性。通过调节橡胶和环氧树脂的熔解度参数,控制凝胶化过程中相分离形成的海岛结构.以分散相存在的橡胶粒子中止裂纹、分枝裂纹、诱导剪切变形,从而提高环氧树脂的断裂韧性。目前用液体橡胶增韧环氧树脂的研究有两种趋势。一种是继续采用CTBN增韧环氧树脂体系,重点放在增韧机理的深入探讨;另一种是采用其它的合适的液体橡胶,如硅橡胶、聚丁二烯橡胶、端羧基聚醚、聚氨酯液体橡胶、聚硫橡胶、含氟弹性体、氯丁橡胶、丁腈橡胶、丙烯酸丁酯橡胶等。
1.2、氨基封端的改性硅橡胶增韧
硅橡胶具有较低的玻璃化转变温度、优良的耐热性和韧性,但直接用于增韧环氧树脂效果不太理想,主要是由于环氧树脂与硅橡胶的溶解度参数相差较大,硅橡胶难以在环氧树脂中有效分散。
改性硅橡胶对环氧树脂具有良好的增韧效果。在硅橡胶分子链上引入活性较强的端氨基.然后对环氧树脂进行改性,在环氧树脂固化过程中,硅橡胶分子链上的端氨基可与环氧基团发生化学反应.因此增进了硅橡胶与环氧树脂的相容性,达到增韧的目的。通过对增韧体的冲击强度测试表明,每百份EP加入硅橡胶0—15份,增韧体的冲击强度随着改性硅橡胶的加入量升高而明显增大。加入量超过15份后,冲击强度增势缓慢。
为了提高硅氧烷与环氧树脂的相容性.加入硅氧烷一环氧基嵌段共聚物,可使硅氧烷均匀、稳定地分散在环氧基体中。采用芳香硅氧烷嵌段共聚物作为相容剂可提高EP的韧性,改性环氧体系的断裂韧性最大值为未改性环氧体系的2倍。人们利用硅氧烷一环氧化合物的嵌段共聚物与环氧树脂共混,使得硅树脂均匀分散于环氧树脂基体中.促进相互间渗透,当分散相粒径小于0.1p,m时,环氧树脂冲击强度提高了l.5倍。
含羟基的线型聚有机硅氧烷,与环氧树脂在160℃进行预反应,生成嵌段共聚物,再加入固化剂交联固化。在环氧树脂与有机硅树脂的混合物中.加入氨基硅烷偶联剂,如KH一550、ND一42等,使氨基和烷氧基与环氧树脂的环氧基和聚硅氧烷的羟基反应生成了嵌段共聚物,提高了二者的相容性。有机硅改性环氧树脂胶粘剂具有很高的耐热性、耐水性和耐老化性。由聚硫橡胶改性环氧溶液为成膜物质,加入金属氧化物填料,添加有机胺固化剂所组成的双组分涂料。该涂料对金属、非金属的附着力强(对钢铁附着力为3-4MPa,对混凝土附着力为4~5MPa)、涂膜坚硬、光滑、丰满,不吸附污浊和藻类,具有韧性好、高弹性、耐候、耐霉菌、耐磨、耐酸碱和耐多种溶剂等特点。
1.3、核壳结构聚合物增韧
核一壳聚合物是一种具有独特结构的聚合物复合粒子,一般采用分步乳液聚合制得。其核为橡胶,赋予制品拉伸性能,壳为具有较高玻璃化温度的塑料,主要功能是使微粒相互隔离。促进在基体中的分散及增加与基体树脂间的相互作用。
核壳结构聚合物fCore—Shell Latex Polymer.CSLP)粒子的内部和外部分别富集不同成分.显示出特殊的双层或者多层结构,核与壳分别具有不同功能,通过控制粒子尺寸及CSLP组成改性环氧树脂,可以获得显著增韧效果。与传统橡胶增韧方法相比,不容性的CSLP与环氧树脂共混,在取得好的效果同时T9基本保持不变,而利用相容性的CSLP则可获得更好的结果。用核壳聚合物改性环氧树脂粘合剂能减少内应力,提高粘结强度和冲击性。如在环氧树脂中分别加入聚丙烯酸丁酯橡胶粒子和PBA/PMMA(聚丙烯酯丁酯/聚甲基丙烯酸甲酯)核壳胶粒,以双氰胺为固化剂所得固化物的结构形态和性能。结果表明,用丙烯酸橡胶粒子可提高环氧树脂的断裂韧性,但远远低于核壳粒子(PBA/PMMA)的增韧效果;在环氧树脂固化过程中,由于PMMA与环氧树脂的相容性好.环氧树脂渗入壳的表层与胶粒发生键合,围绕核壳粒子的环氧基体由于塑性变形,能量吸收水平增加,断裂韧性大幅度提高。
加入30份PnBA/PMMA核壳结构增韧剂后,环氧树脂的冲击强度有显著提高,断裂方式由脆性断裂转为韧性断裂。对于酸酐固化体系.冲击强度提高约32倍,超过ABS等工程塑料,对于Moca固化体系,冲击强度提高近7倍。对比就地聚合PBA—P(BA—IG)0.2一l p。m的橡胶粒子分散体以及用晶种乳液聚合制成的PBA/PMMA,P(BA—IG)/P
(MMA—IG)橡胶粒子分散体分别在环氧树脂体系中的内应力减低效果。发现前者固化产物T9下降了,而后者T9完全没有影响。SEM观察,前者形成了IPN结构,而后者仅仅是粒子界面附近形成IPN.同时后者制成的粘合剂的性能有了明显的提高。就地聚合获得的第一代丙烯酸橡胶粒子其核壳结构基本上是均一的,它们作为结构胶,其剥离强度、冲击性能还不很好。晶种核壳聚丙烯酸橡胶粒子是第二代产品,其薄壳部分具有絮凝性.核部分担负着增强韧性作用。研究中发现,后者环氧树脂固化后核部分的丙烯酸橡胶粒子呈微分散型,因此抗冲击性、剥离强度较高。

1.4、端硅烷基聚醚增韧
为了制得既具有高粘接强度又兼备抗剥离、防震动、耐疲劳,在室温下又可固化的胶粘剂.选择合适的端硅烷基聚醚母体树脂与环氧树脂相互配合配制出无溶剂反应型液体弹性胶粘剂。端硅烷基聚醚主链结构属大分子纯醚链结构,因而其具有良好的柔曲性,高延伸性和耐水解性,其粘度明显降低.因此所制得的密封胶可不用溶剂.不用或少用增塑剂即获得良好的工艺操作性,所带的烷氧基甲硅烷基团与空气中的湿气接触后通过水解一缩合反应形成Si—O—Si键,赋予体系耐侯、耐水、耐老化和耐久的优良性能。
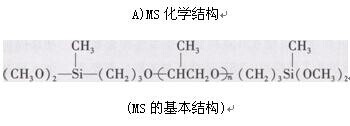
B)合成路线
第一步:以烯丙基聚醚为原料。以二氯甲烷为扩链剂在苛性碱催化剂存在下,通过扩链反应制得烯丙基封端的聚醚中间体。
第二步:精制的中间体进行端硅烷基化反应.制得端硅烷基聚醚。立即在Pt Cat,使中间体与甲基二甲氧基硅烷进行硅氢加成反应,制得MS聚合物。

C)固化机理
MS聚合物的固化机理根据硅氧烷基固化有机硅的机理。发生两部反应:硅烷基团的水解.然后发生缩合反应,有机锡可催化固化。
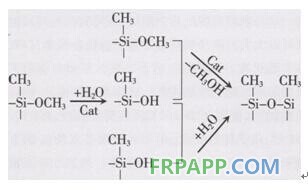
在化合物中MS一聚合物的交联反应速度取决于以下两个因素:空气的湿度和工作环境的温度适宜。成膜速度和完全固化速度随温度升高而加快,在低温及低相对湿度的冬季。固化反应速度也就减慢。MS与环氧树脂配合,可形成高性能胶粘剂。用于不同基材的弹性粘接。
在端硅烷基聚醚一环氧型胶粘剂体系中,液体双酚A二缩水甘油醚(液体双酚A环氧树脂)主要是对端硅烷基聚醚橡胶母体起补强作用。这种补强作用是通过以下途径实现的。
(1)在硫化后的端硅烷基聚醚一环氧体系中,环氧树脂的微粒子于MS聚合物母体获得高效的补强作用。
(2)在胶粘剂体系中采用分子中含有NH2基的端硅烷基聚氧化丙烯,它有助于使橡胶母体与环氧树脂通过Si一0一Si键和一NHCH2CHCH2一键交联成为一个三维网络整体。
(3)在体系中采用单氨基或双氨基烷氧基硅烷,如N—B一氨乙基一r一氨丙基三甲氧基硅烷或-r一氨丙基三乙基硅烷等,也可以通过形成上述b中所述的良种化学键将橡胶母体和环氧树脂桥联成一个三维网络结构。
端硅烷基聚醚一环氧型胶粘剂的主要特性归纳如下:
(1)在无溶剂的情况下体系粘度低,流动性好.工艺操作性能优良。
(2)在常温下可于短期内固化,表干时间短。具有良好的室温硫化性能。硫化后产物不仅具有优良的柔曲性和弹性,而且还具有很低的收缩率且无应力集中现象。
(3)硬化成膜的弹性体呈橡胶态.具有优异的冲击强度和剥离强度。体系的T型剥离强度一般情况下可在100~150N/25mm的范围内,有的特用配方可高达200N/25mm以上。此外,体系的耐振动疲劳性能优良。
(4)在一30—120℃的温度范围内,具有良好的强度保持性,即使在I CE时,其T型剥离强度仍保持20N/25mm,拉伸剪切强度仍为4MPa。
(5)强度性能对测试速度的依赖性小,或者说.强度性能的变化受测试速度的影响不敏感。
(6)胶粘剂对从金属到塑料等广阔被粘基材.即使不经特殊的表面处理也能获得良好的粘附性能。呈现出对基材具有良好的适应性。
2、热塑性工程塑料增韧
热塑性树脂增韧环氧树脂的机理和橡胶增韧环氧树脂的机理没有实质性差别,一般仍可用孔洞剪切屈服理论或颗粒撕裂吸收能量理论。但是从实验结果看,热塑性树脂增韧环氧树脂时。基体对增韧效果影响较小,而分散相热塑性树脂颗粒对增韧的贡献起着主导作用。一般发生桥联约束效应和裂纹钉锚效应。
(1)桥联约束效应与弹性体不同,热塑性树脂常具有与环氧基体相当的弹性模量和远大于基饰的断裂伸长率,这使得桥联在已开裂脆性环氧基体表面的延性热塑性颗粒对裂纹扩展起约束闭合作用。
(2)裂纹钉锚效应颗粒桥联不仅对裂纹前缘的整体推进起约束限制作用,分布的桥联力还对桥联点处的裂纹起钉锚作用,从而使裂纹前缘呈波浪形的弓出状。
用热塑性树脂与环氧树脂形成高分子合金来增韧改型的研究工作,弥补了橡胶弹性体改性环氧树脂的不足。热塑性树脂增韧环氧树脂的研究始于20世纪80年代,使用较多的有聚醚砜(PESl、聚砜(PSD、聚酰亚胺(PEI)、聚醚醚酮(PEEK)、聚苯醚等热塑性工程塑料。目前较新的成果有:用热塑性树脂混合物改性的环氧树脂,改性剂用的是聚砜(Udel Pl700)和聚醚酰亚胺(Ultem l000)的混合物,改性后的环氧树脂用新型芳香二胺固化后T9很高、吸水率降低、耐湿热性能改善很大:用芳香族聚酯改性环氧树脂,双酚A型环氧树脂Epikote828随聚酯分子质量的增大破坏韧性值在增大,但分子质量大到一定程度反而会下降,聚1,4一丁二醇的分子质量为1000时制得的聚酯,添加量仅5%就可使Epikote828体系的伸长率提高50%,拉伸强度提高25%;用不同胺封端的芳醚酮齐聚物增韧.随着齐聚物量的增加断裂能提高但玻璃化温度下降,在交联固化时相的分离是韧性提高的根源,这些树脂的结构取决于增韧剂的用量。
采用聚醚酰亚胺提高双酚A二氰酸酯/酚醛环氧树脂共混物的断裂韧性。实验结果表明,聚醚酰亚胺是氰酸酯/酚醛环氧树脂共混物的有效增韧剂,加入l5%的聚醚酰亚胺可使断裂韧性提高到1.45MPa,弯曲强度也有所提高。用扫描电子显微镜和动态粘弹谱研究改性共混物的微观结构发现有双连续结构,共混物的韧性和耐溶剂性主要与相行为有关,固化工艺对含l0%聚醚酰亚胺的共混物的断裂韧性和形态没有明显的作用。不同含量的聚酰亚胺醚(PEI)对TGDDM/DDS体系的增韧效果。PEI含量为15wt%时,树脂的GIC高达0.54kJ/n7.比增韧前提高了约2倍。研究PEl含量对环氧和PEl两相形态的影响时发现,当PEl含量小于l5wt%时.PEl以颗粒形式分散到环氧连续相中:PEl含量高于15wt%后,体系发生相转变,部分PEI形成连续相,随PEl含量的继续升高,PEl变为连续相(30wt%),环氧变为颗粒状分散相,树脂的韧性显著提高。
将胺化聚碳酸酯(a—PC)和环氧树脂fEPl以一定比例混合,加热到l20~160℃后加入熔化的二氨基二苯基甲烷,制备固化的胺化聚碳酸酯增韧环氧树脂。结果表明,EP与a—PC形成了网络结构.且当a—PC质量分数为10%时,试样断裂韧性最大.
双马来酰亚胺耐热性能好,利用其改性环氧树脂可以大大提高环氧树脂高温下的粘合强度。以双马来酰亚胺、环氧树指、芳香二胺为原料制备出了新型的环氧树脂增韧体系。该体系耐热性好、粘合性能优异,室温下及200℃测其剪切强度(45#钢/45#钢)几乎没有变化,用加入烯丙基双酚A的方法来增加环氧树脂与BMI相容性。通过红外光谱分析发现烯丙基双酚A可与双马来酰亚胺发生接枝共聚反应,形成带有环氧基团的双马来酰亚胺树脂,在加入固化剂时可与环氧树脂发生固化交联,使体系中的两相具有良好相容性。得到一种耐高温的韧性环氧改性树脂。以环氧树脂为基础合成了环氧双马来酰亚胺(EB)。该体系由功能性双马来酰亚胺与环氧树脂反应而成,固化则利用双马来酰亚胺的固化机理。该体系不仅具有环氧树脂的粘接性好、固化收缩率低的特点。而且还具有类似双马来酰亚胺树脂的高耐热性。同时,该体系的冲击性能也比双马来酰亚胺有了较大的提高。
聚酰胺酸(PAA)是聚酰亚胺(PI)的反应中间体。与PI相比,PAA在低沸点溶剂中即可制得。PAA改性环氧树脂体系与Pl改性体系相比较具有更加优异的剥离性能。利用PAA改性环氧树脂时,其自身相当于环氧树脂的固化剂,可以与环氧基团形成类酯结构,同时,PAA本身又具有一定的活性.可以酰胺化形成Pl长链,使固化体系表现出高的粘结剪切强度和耐热性能。在THF/CH30H混合溶剂中利用PMDA与ODA合成出PAA并成功地用作环氧树脂的固化剂和改性剂。改性体系由于PAA与EP之间的协同作用而具有良好的综合性能。同时该体系固化时低沸点溶剂易于挥发,不会造成大的内应力。由于材料中的内应力通常是造成材料综合性能下降的原因。他们采用两阶段固化工艺来充分排除固化体系中残存的溶剂和气泡以进一步提高体系的综合性能。
近些年,高强轻质纤维增强复合材料在超低温环境中逐渐使用,对环氧树脂的超低温性能研究也日益加强。在作为复合材料液氢贮箱的基体材料以及在超导领域中用作胶粘剂,浸渍料和纤维增强复合材料的基体材料等方面,我国的研究已取得一些进展。纯环氧树脂具有很高的交联密度,即使在常温下也存在着质脆、韧性低、抗冲击性差等缺点。而作为复合材料的树脂基体,一般都需要在很高的温度下固化。在固化后冷却过程中,由于热收缩树脂基体内部会产生热应力,当温度从室温降低至超低温(-150℃以下)时,基体内因热收缩而产生的内应力将更加显著,而一旦热应力超过树脂本身的强度,就会导致树脂基体的破坏。因此,提高韧性对环氧树脂在超低温下的使用至关重要。
目前提高环氧树脂超低温韧性的方法主要是使用柔性的脂肪族树脂(如PPGE)和液体橡胶以及柔性固化剂(如POPDA)来增韧环氧树脂。由于此类材料玻璃化转变温度较低,常温下具有较大的自由体积,当温度降至超低温时,树脂体系会产生很大的热收缩,导致较大的热应力,这限制了其在超低温下的应用。
目前一个重要的研究方向是,常温下高性能热塑性塑料与环氧树脂的共混改性,可使共混体系同时兼具有两者的优越性能,即在保持热固性树脂高模量的同时,又兼具热塑性塑料的高韧性。迄今为止,国内外对热塑性塑料能否在超低温下增韧环氧树脂及其如何影响树脂体系的力学性能这一问题正在作深入系统的研究。与柔性链增韧体系相比.热塑性塑料增韧环氧树脂体系具有较高T9,常温下的自由体积很小,因而可以减小从室温冷却至超低温过程中产生的热应力。所以,研究热塑性塑料增韧环氧树脂体系在超低温下的性能,对提高热固性树脂在超低温领域的应用有重要意义。
用一种新型含氮杂萘酮结构的聚醚腈酮(PPENK)及其与环氧聚醚的混合体系增韧环氧树脂,测试了两类增韧体系在室温(Rn和液氮温度(LND下的断裂韧性(Kic)和冲击强度,并研究了PPENK对环氧树脂体系在室温和超低温下力学性能的影响。研究表明,虽然聚醚对增加PPENK改性环氧树脂体系的室温韧性贡献不大,但却明显提高了其超低温韧性。PPENK的引入对环氧树脂在室温和液氮温度下的力学性能影响不同,室温下PPENK的引入使强度下降而模量不变或略有增加,而液氮温度下对弯曲及压缩强度影响较小、拉伸强度则下降了32.8%,拉伸模量下降显著f降低了37.5%)。同时,PPENK的引入使室温和液氮温度下的断裂延伸率都有所下降。
3、液晶高分子增韧
液晶高分子聚合物是一类分子中含有液晶单元的高分子化合物。通常按其形成液晶态的物理条件可分为溶致型液晶和热致型液晶。利用热致型液晶TCIP增韧环氧树脂既能提高其韧性,又能确保不降低环氧树脂的其它力学性能和耐热性。热致性液晶聚合物增韧环氧树脂已成为环氧树脂重要的增韧改性方法。液晶聚合物(LCP)含有大量的刚性介晶单元和一定量的柔性间隔段,其结构特点决定了它的优异性能,它比一般聚合物具有更高的物理力学性能和耐热性。TCLP增韧机理主要是裂纹钉锚作用机制,(TCLP)作为第二相(刚性与基体相近),本身有一定的韧性和较高的断裂延伸率,因此只需少量就能增韧环氧树脂,同时提高其模量和耐热性。TLCP比其它聚合物具有更高的物理力学性能和耐热性,它在加工过程中受到剪切作用,形成纤维结构,具有高度自增强作用。TLCP改性环氧树脂固化后体系为两相结构,TLCP以原纤的形式存在于体系中,可阻止裂缝,提高基体韧性.而材料的耐热性和刚度不降低或有所提高,它的拉伸强度可达200MPa以上,比ET、PC高3倍,比PE高6倍;其模量达20GPa以上,比PE高20倍,比PC、PEK高8.5倍。LCP还有另一个重要特点:它在加工过程中受到剪切力作用具有形成纤维状结构的特性.因而能产生高度自增强作用。因此当用热致性液晶聚合物(TLCP)和环氧树脂进行共混改性时,在提高韧性的同时,弯曲模量保持不变、T9还略有升高,同化物为2相结构。LCP以原纤形式分散于环氧基体中,在应力作用下提高了材料的韧性。LCP和热塑性工程塑料相比用量仅为其25~30%,却可达到同样的增韧效果。一种侧链高分子液晶LCGMB来增韧环氧基体,该化合物在增韧环氧树脂时,柔性的液晶分子主链能弥补环氧基体的脆性,侧链的刚性单元又保证了改性体系的模量不会下降,从而提高体系的综合力学性能。在研究时还发现体系的冲击性能随LCGMB的用量增大而增大,当用量为20%~30%摩尔分数时有最大冲击性能。经SEM观察分析,其冲击断口环氧树脂呈连续相,液晶则以微粒形式分散在树脂基体中。当受到冲击时液晶微粒是应力集中源并诱发周围环氧基体产生塑性形变吸收能量。
用含有芳酯的液晶环氧4,4’一二缩水甘油醚基二苯基酰氧(PHBHQ)t曾韧E一51环氧树脂.选择熔点与液晶相玻璃化温度相一致.反应活性较低的混合芳香胺为固化剂,当PHBHQ的质量分数达50%时固化树脂冲击强度为40.2J/m2.与不加PHBHQ的冲击性能相比较,提高31.72J/m2,此外玻璃化温度也有一定的提高。一种端基含有活性基团的热致性液晶聚合物(LCPUl,用其改性环氧树脂CYD一128,4`一二氨基二苯砜(DDSl固化体系,对改性体系的冲击性能、拉伸性能、弹性模量、断裂伸长率、玻璃化转变温度T9与LCPU含量的关系进行了探讨,将不同种类液晶化合物对CYD一128/DDS体系改性效果进行了比较,用扫描电镜(SEM)对材料断面的形态结构进行了研究。结果表明.LCPU的加入可以使固化体系的冲击强度提高2~3.5倍,拉伸强度提高l.6~1.8倍,弹性模量提高1.1~1.5倍,断裂伸长率提高2~2.6倍,T9提高36。60℃,改性后材料断裂面的形态逐渐呈现韧性断裂特征。一种侧链型液晶聚合物(SLCP),用T31作固化剂时SLCP对环氧树脂有较好的增韧效果。在强度和玻璃化温度不降低的情况下,断裂伸长率比未改性固化物最大提高2.6倍,但用三乙醇胺作固化剂时SLCP对环氧树脂改性效果不明显。
用含有芳酯介晶单元的液晶环氧4、4一二缩水甘油醚基二苯基酰氧(PHBHQ)增韧E一51环氧.选择熔点与PHBHQ介晶相温度相一致、反应活性较低的混合芳香胺为固化剂.当PHBHQ用量达50wt%时,固化树脂的冲击强度达40.2kJ/rd,与不加PHBH0时鲥冲击强度23.0kJ/前相比较。提高了近2倍。此外玻璃化温度也有一定提高。
4、互穿网络聚合物增韧
IPN增韧改性方法互穿聚合物网络(IPN)是制备特殊性能的高分子合金的有效方法。IPN是由两种或两种以上组成和构型不同的均聚物或共聚物相互贯穿,缠结形成的聚合物混合物,是特殊的多相体系。其特点是一种材料无规则地贯穿到另一种材料中,使得IPN体系中两组分之间产生了协同效应,起着“强迫包容”作用,从而产生出比一般共混物更加优异的性能。
影响IPN性能的主要因素有网络的互穿程度、组分比、交联程度,全互穿IPN明显高于半互穿IPN的性能。IPN的橡胶相组分过大。抗拉强度、抗剪切强度、抗弯曲强度都急剧降低,增韧效果也差。适当的交联都可获得最佳力学性能.不但韧性大幅度提高。而且抗张强度也有所提高。但交联含量过高,对提高固化物韧性不利,因为网络链太短,不利于外力作用下的应变,吸收冲击能减小。
互穿网络聚合物增韧环氧树脂国内外进行了大量的研究,其中包括:环氧树脂一丙烯酸酯体系、环氧树脂一聚氨酯体系、环氧树脂一酚醛树脂体系和环氧树脂一聚苯硫醚体系等,增韧效果满意。主要表现在环氧树脂增韧后不但冲击强度提高,而且拉伸强度不降低或略有提高,这是一般增韧技术无法做到的。同步法制造环氧树11/聚氨酉I(EP/PUR)IPN、原位聚合法制备刚性PU改性环氧树脂、同步法合成聚丙烯酸正丁11/环氧树I旨(PnBA/EP)、用环氧树脂和双酚A为聚合单体制备高分子质量环氧树脂等成果,都有很好效果。
4.1丙烯酸增韧改性环氧树脂
利用丙烯酸类物质增韧环氧树脂可以在丙烯酸酯共聚物上引入活性基团,利用活性基团与环氧树脂的环氧基团或羟基反应,形成接枝共聚物,增加两相间的相容性。另一种方法是利用丙烯酸酯弹性粒子作增韧剂来降低环氧树脂的内应力。还可以将丙烯酸酯交联成网络结构后与环氧树脂组成互穿网络(IPN)结构来达到增韧的目的。
利用环氧树脂与甲基丙烯酸加成聚合得到环氧一甲基丙烯酸树脂(EAM),其工艺性与不饱和聚酯相似,化学结构又与环氧树脂相似,得到的改性树脂体系经固化后不仅具有优异的粘合性和化学稳定性。而且具有耐热性好、较高的延伸率,固化工艺简单等优点。同时由于共聚链段甲基丙烯酸酯的引入,体系固化时的交联密度降低,侧基的引入又为主链分子的运动提供更多的自由体积,因此改性体系的冲击性能得以提高。
将线性聚丙烯丁酯交联成网状结构后与环氧树脂及固化剂固化,形成互穿网络结构。该方法增加了丙烯酸丁酯与环氧树脂的相容性。该互穿网络体系具有较高的粘接强度和优异的抗湿热老化能力。聚丙烯酸正丁酯/环氧树脂(PnBA/EP)IPN,与纯环氧树脂相比,使用不同固化剂,其冲击强度可提高20%~200%,当加入10%PnBA时,其弯曲强度和模量都有所提高,而且挠度增加,IPN试件耐热性能有所下降。固化剂的选择以及PnBA量的控制是得到最佳IPN性能的影响因素,采用优化工艺合成环氧树脂一丙烯酸酯树脂的混合物乳液,IPN有助于材料的玻璃化温度强和热分解温度Td的提高。用丙烯酸酯封端的聚硫橡胶(Acry—LP)和环氧封端的聚硫橡胶(EP—LP)与双酚A环氧树脂(EP一51)合成互穿网络和共聚网络聚合物,这两种体系都具有低温柔顺性和良好的力学性能。
4.2聚氨酯增韧环氧树脂
用聚氨酯改性环氧树脂主要是为了改善其脆性,提高其柔韧性,增加剥离强度。聚氨酯粘接性能好.分子链柔顺,在常温下表现出高弹性。利用高分子合金的思想,采用熔体共混法制备出了PU/EP共混体系。以异氰酸根封端的聚氨酯预聚体与环氧树脂在熔融条件下加入固化剂固化后得到共混改性体系:由于异氰酸根本身能与环氧基团反应,因此得到的改性体系两相间有良好的相容性,利用DMA分析.可发现其谱图上在m(PU):m(EP)=20:80时只有单一的宽的玻璃化转变蜂,这进一步证明了两相间的相容性。改性体系比环氧树脂的冲击强度有了大幅度提高。
目前研究最多的聚氨酯增韧环氧树脂体系是以聚氨酯与环氧树脂形成SIPN和IPN结构,这两种结构可起“强迫互容”和“协同效应”作用,使聚氨酯的高弹性与环氧树脂的良好的耐热性、粘接性有机地结合在一起,取得满意的增韧效果。利用双酚A环氧树脂与末端为异氰酸酯的聚醚聚氨酯低聚物进行改性接枝.然后在DDM固化剂作用下形成线性聚氨酯贯穿于环氧网络的半互穿网络结构。两者在用量比为70/30时有很好的协同性能。体系的剪切、剥离强度与冲击强度均有较大程度的提高,体系的断裂延伸率由环氧基体的2.09%提升至211.9%,断裂强度提高了18.56MPa,同时该体系还具有良好的阻尼特性。PU/EP的相行为与粘接剪切性能的关系,通过红外光谱分析发现,该体系中不仅存在着EP与PU的各自的交联反应,还存在二者的共聚反应。用DSC对其进行分析发现该体系在高温下有单一宽的玻璃化温度,同时还发现体系的玻璃化温度随环氧树脂用量增加而提高,甚至高于EP基体强,其原因是EP用量增大后,PU与EP的接枝反应增多,分子间作用力增大,从动态力学谱图上也可看出,损耗峰向高温方向移动。通过TEM观察发现,体系两相间界面模糊,这进一步证明了两相间的相容性。体系中存在的聚氨酯与环氧树脂的接技共聚物大大增加了二者的互穿效应.从而体系的综合性能得以提高。
利用高分子合金的思想,采用熔体共混法制备出了PU/EP共混体系,原理是以异氰酸根封端的聚氨酯预聚体与环氧树脂在熔融条件下加入固化剂固化后得到共混改性体系。由于异氰酸根本身能与环氧基团反应,因此得到的改性体系两相间有良好的相容性,利用DMA分析可发现只有单一的宽的玻璃化转变蜂,这进一步证明了两相间的相容性,这使改性体系冲击强度有了大幅度提高。
橡胶弹性体的加入使环氧树脂的韧性大幅度提高,是以牺牲耐热性和刚性为代价的,而且对高交联密度的环氧树脂,橡胶弹性体的增韧作用非常小;用原位聚合法制备刚性聚氨酯大分子来改性环氧树脂。当固化体系中刚性聚氨酯含量不大时.刚性分子能以分子水平均匀分散于环氧基体中形成分子复合材料,整个体系类似于半一互穿网络。这些刚性分子能对基体起到增强作用,提高基体拉伸强度,同时又能阻止裂纹而增大基体的韧性。
5、刚性高分子增韧
刚性高分子增韧环氧树脂正日益受到重视.采用原位聚合技术使初生态刚性高分子均匀分散于刚性树脂基体中,得到准分子水平上的复合增韧特性是探索改性脆性高聚物,得到高强度和高韧性聚合物的新途径。原位增韧是通过两阶段反应,使在交联后形成分子量呈双峰分布的热固性树脂交联网络,这种方法制得树脂韧性可以是常规树脂韧性的2-10倍。其增韧机理可能是由于形成的固化物交联网的不均一性,从而形成了微观上的非均匀连续结构来实现的。这种结构从力学上讲有利于材料产生塑性变形,所以具有较好的韧性。
原位聚合苯甲酰胺(PNM)对环氧树脂和粒子填充环氧树脂的改性作用研究表明:加入5%左右的。NM,环氧树脂拉伸强度从纯环氧树脂的50.91MPa和粒子填充(30%)环氧树脂的69.21MPa.分别提高到94.25MPa和91.85MPa:断裂韧性从纯环氧树脂的0.83J/m2和粒子填充环氧树脂的0.72J/m2,分别提高到l.86J/m2和1.98J/mz,而其他性能也有不同程度的改善。原位增韧是通过两阶段反应.使在交联后形成分子质量呈双峰分布的热固性树脂交联网络,这种方法制得树脂韧性可以是常规树脂韧性的2~10倍。其增韧机理可能是由于形成的固化物交联网的不均一性,从而形成了微观上的非均匀连续结构来实现的这种结构从力学讲有利于材料产生塑性变形,所以具有较好的韧性。
6、纳米粒子增韧环氧树脂
纳米粒子增韧环氧树脂是环氧树脂改性研究的新领域。纳米粒子尺寸界定在1~100nm之间.具有极高的比表面积,表面原子具有极高的不饱和性,因此纳米粒子的表面活性非常大。在利用纳米粒子增韧环氧树脂时,环氧基团在界面上与纳米粒子形成远大于范德华力的作用力,形成非常理想的界面,能起到很好的引发微裂纹、吸收能量的作用。利用分散剂可实现纳米粒子与环氧树脂的均匀混合解决了纳米粒子由于粒径过小易团聚的问题.两者在界面处存在着较强的分子间力,因此有较好的相容性,而且纳米粒子以第二聚集体的形式较均匀地分散在树脂基体中,使两者粘接性能好.因而在受冲击时能起到吸收冲击能量的作用,从而达到增韧的目的。纳米Si02粒子可使环氧树脂的冲击性能和拉伸性能大幅度提高,在改性体系中纳米粒子呈分散相,环氧树脂为连续相。利用SEM观察纯EP冲击断口与ElY/粘土纳米复材冲击断口时发现,前者断口为光滑脆性断裂形貌特征,而后者断E1则凸凹不平,表现出韧性断裂形貌特征。其原因为纳米刚性粒子在复材体系中作为应力集中物在受力时既能引发银纹,又能终止银纹。同时由于纳米粒子具有强的刚性,裂纹在扩展遇到纳米粒子时发生转向或偏转吸收能量达到增初之目的。纳米SiO2粒子可使环氧树脂的冲击性能和拉伸性能大幅度提高。
7、柔性链段固化剂增韧环氧树脂的研究
含有柔性链段的大分子固化剂增韧环氧树脂.其柔性链段能键合到致密的环氧树脂交联网络中.并在固化过程中产生了微观相分离,形成了致密、疏松相间的两相网络结构,在提高环氧树脂韧性的同时,又简化了成型工艺。利用具有柔性链的双羟基化合物中听含的羟基与环氧树脂的环氧基进行反应,将柔性链段引入到环氧主链中,制得低粘度的环氧树脂,再用丙烯酸酯化,可得到紫外光固化的低粘度的环氧丙稀酸酯涂料。含聚乙二醇(PEGl柔性间隔基的扩链脲可用来增韧改性环氧E一51/二苄胺(DBA)体系。当脲分子中PEG分子量为6009/mol时,环氧E一51/扩裢脲/DBA体系固化物抗冲击强度最高可达55.8kJ/nf,约为环氧E一51/DBA固化体系的5倍。
国外合成了一系列用于增韧环氧树脂的端氨基芳醚酮固化剂,如端氨基聚双酚A醚二苯酮(BPAPK),端氨基聚3异丁基对苯二酚醚二苯酮(tBPK)等。该固化剂固化的环氧树脂由于醚网络的存在,提高了交联点间链段的柔软性和扭转特性.从而使固化物呈现高韧性。用分子量为70009/mol的BPAPK增韧改性环氧Epon828/DDS体系.当BPAPK的含量为40wt%H寸,材料的断裂能达300J/前,与增韧前的300J/rff相比,断裂能提高了6倍多。而常用的聚酰胺650、651和聚醚胺230、430、2000也都是不错的选择。







 鲁ICP备2021047099号
鲁ICP备2021047099号